Hundreds of thousands of people die from malaria each year, but it took 141 years to develop a vaccine for it. Advance market commitments could speed things up next time.
In 1969, just 14 years after it was initiated, the World Health Assembly stepped back from its program to eradicate malaria worldwide, declaring that it was no longer feasible in the near future.
Global eradication was an ambition that had been galvanized by the promise of DDT, a pesticide developed during World War Two. The program’s leaders expected malaria eradication to be a smooth, quick process – one that would be completed within just eight years in some countries like Mexico, and 10 to 15 years worldwide.

Subscribe for $100 to receive six beautiful issues per year.
But 15 years later, the end was still distant. The strategy was re-examined in 1969, and then effectively suspended.
In the regions where eradication does not yet seem feasible, control of malaria with the means available should be encouraged and may be regarded as a necessary and valid step towards the ultimate goal of eradication.
– World Health Assembly, 1969.
The story of malaria’s failed eradication campaign ran alongside another story – the long drawn-out struggle to develop a malaria vaccine.
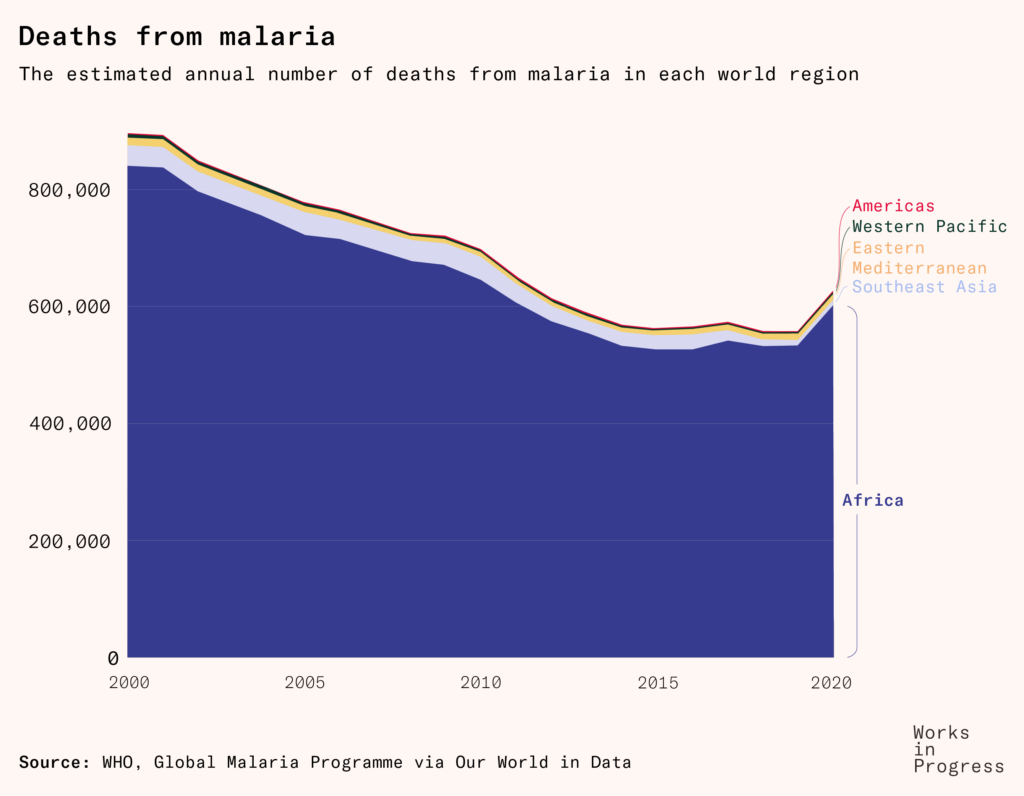
The question of why we didn’t get a malaria vaccine sooner isn’t just an intellectual exercise – around 600,000 people die from the disease each year – and its answer isn’t just a scientific one.
The malaria parasite is complex, making it much more difficult to develop a vaccine than usual. But at the heart of the issue, especially in recent decades, was a lack of financial incentive and urgency. Malaria primarily affects the global poor, whose ability to spend on healthcare is limited. Companies who invent solutions for the poor face pressure to keep prices so low that it is hard to profit, on top of the fact that vaccines tend to make less money than other medicines.
Advance Market Commitments, a way of promising to buy products that don’t yet exist, could help overcome these barriers, encourage investment in vaccines for other diseases, and improve on the vaccines we currently have.
But let’s step back and start at the beginning to see how the story unfolded.
The discovery of the parasite
The germ that caused malaria wasn’t discovered until the late 1870s. Until then, the closest scientists had come was identifying dark pigments in post-mortem tissues from malaria victims, which they attributed to a range of different causes.
Some believed that the pigments were caused by a species of Bacillus bacteria – because two Italian doctors had injected Bacillus into rabbits, and observed their spleens swell, just like in a human malaria infection. They called this species Bacillus malariae. But their claims received a lukewarm reception from other scientists, who were unable to replicate their observations.
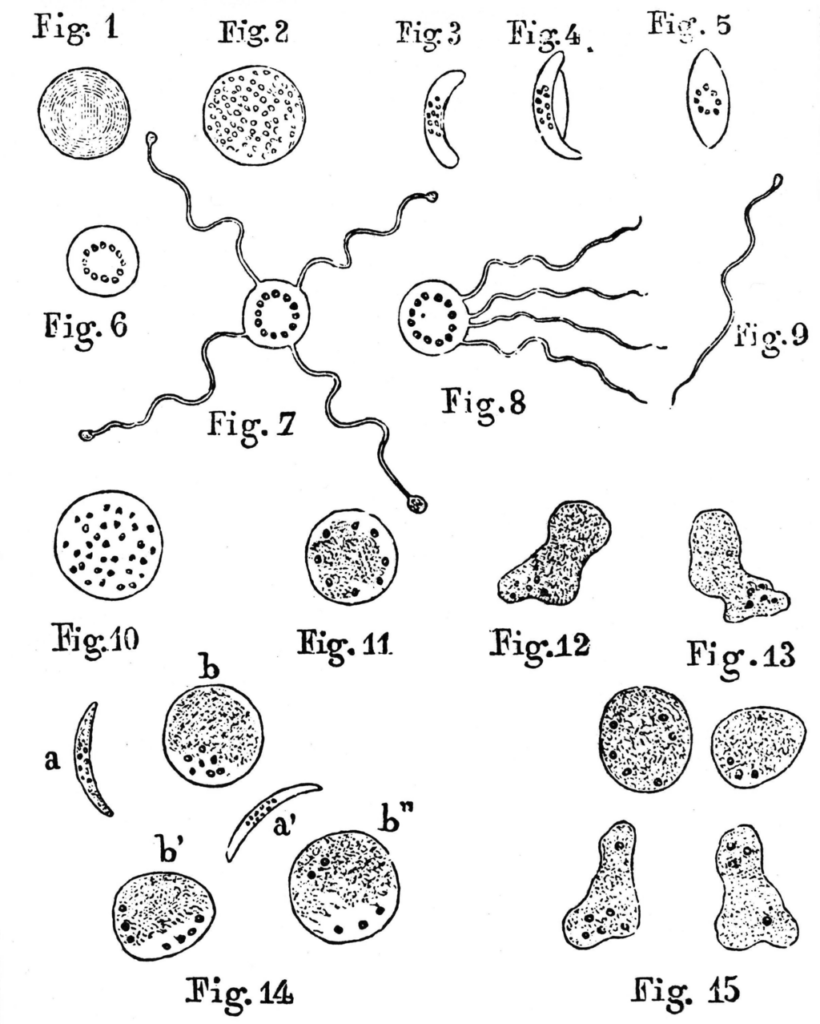
The real breakthrough came in 1878, when a young French doctor called Charles Laveran was posted to a military hospital with malaria patients in Algiers. Like other doctors, he observed the classic dark pigment in post-mortem organs. But he went further by examining wet blood from living patients.
On October the 20th (1880) while I was examining microscopically the blood of a patient (a drop of blood under a coverslip sealed with paraffin) suffering from malaria, I noticed among the red corpuscles, elements that seemed to me to be parasites. Since then I have examined the blood of 44 malaria patients; in 26 cases these elements were present. These elements were not found in the blood of patients who were not ill with malaria.
– Charles Louis Alphonse Laveran, ‘Note sur un nouveau parasite trouvé dans le sang de plusieurs malades atteints de fièvre palustre’, Bulletin Académie Médecine, 9 (1880), 1235–1. As translated in Garnham, 1988. Bolded text our emphasis.
Laveran had discovered that malaria was caused not by a virus or a bacterium, but by a microscopic parasite.
When he published his findings that year, scientists were divided. Robert Koch, one of the most eminent scientists of the time, was simply dismissive. Others believed that rather than parasites, he had simply found byproducts of red blood cells that had broken down.
Laveran persisted. He found the same crescent-shaped bodies containing pigment granules in malaria patients from other hospitals, while finding none in patients with other diseases.
He could see them transform under the microscope with his own eyes, and watched as they grew filaments and became highly motile, visibly alive.
{kind=link}
He continued to write on the topic, and more and more scientists replicated his findings. The parasite became known as Plasmodium, and was settled as malaria’s cause.
The initial skepticism Laveran met wasnʼt entirely unreasonable – it’s not always been easy to identify the pathogen that causes a disease.
Viruses, for example, were too small to be seen with microscopes of the time. Often, patients had multiple infections simultaneously, so scientists might have just identified the wrong pathogen. Or, supposedly healthy control patients might also be carrying the pathogen without symptoms, making it harder to pinpoint as the cause. Or the real pathogen might be difficult to maintain in the lab.
When Edward Jenner for example developed the first ever vaccine in 1796, for smallpox, the underlying reason it worked was still a mystery. He didn’t know it was caused by a microorganism – let alone a virus – because germ theory hadn’t yet been pieced together.
The first virus would be discovered around 90 years later (the tobacco mosaic virus), around the time of the second-ever vaccine (the rabies vaccine).
Laveran’s discovery of the malaria parasite transpired during this new Golden Age of Microbiology.
Evidence for germ theory had grown and grown over the nineteenth century, and was becoming consensus by the time of his discovery. The historical explanation for malaria – that it was spread by a noxious gas around swamps and marshes – was falling out of fashion.
By now, Pasteur, Koch, Löffler and many other scientists had identified a growing number of microorganisms that caused different diseases, and vaccine development had begun to lift off. What had once been a shot in the dark – scientists hoping to isolate the correct pathogen, often without even knowing what it was – was coming to light.
The difficulties of finding a vaccine: animal models
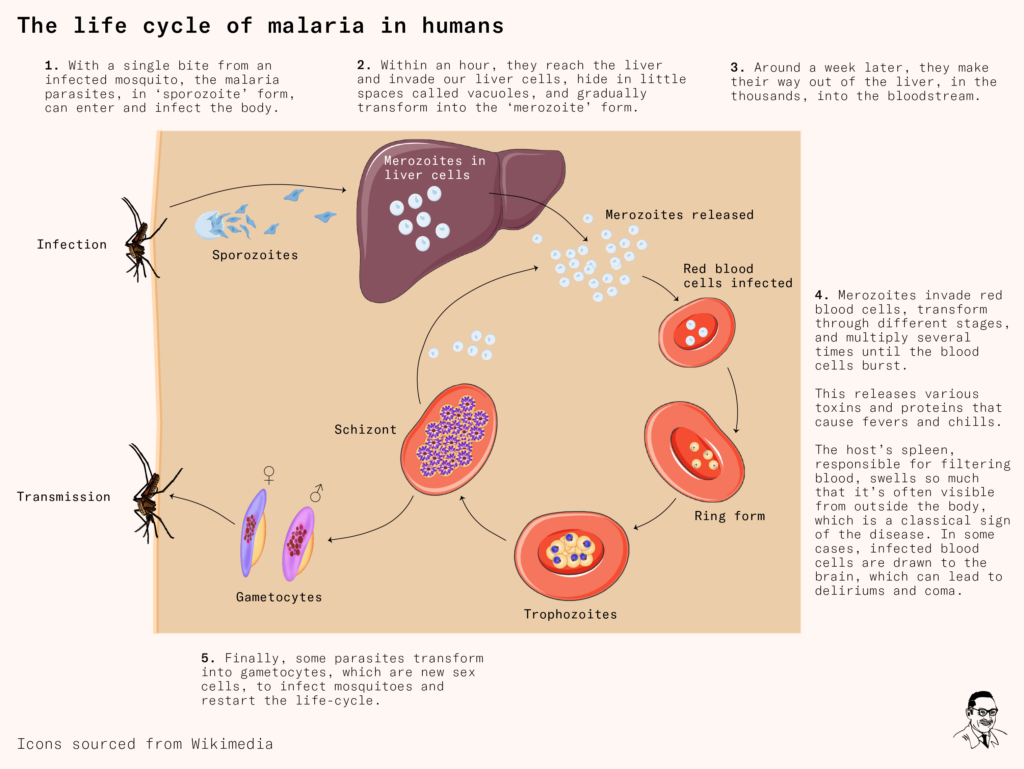
Developing a vaccine for malaria was not going to be easy. As Laveran and others soon realized, the Plasmodium parasite had a very complicated life cycle. It went through several shape-shifting transformations that thwarted the human immune system, and it infected multiple species to complete its life cycle.
After entering our body through a mosquito bite, the parasite – in its first ‘sporozoite’ form – rapidly reaches the liver and invades our liver cells. As we recruit immune cells to attack it, it hides in little spaces called vacuoles where it transforms into another shape.
Around a week later, it’s multiplied into the thousands. The parasites now make their way out of the liver and into our red blood cells. There, they transform several times, consume hemoglobin, and multiply rapidly, before bursting our red blood cells open. This leads to many classic symptoms of the disease – repeated fevers and chills, sweating, anemia and jaundice.
Finally, some parasites turn into ‘gametocytes’ (sex cells) and are sucked up by another mosquito, restarting the life cycle in another victim.
All this complexity wouldn’t be the only obstacle to developing a vaccine, but it would be a recurring theme.
Over the following twenty years, scientists established that malaria was spread to humans by female Anopheles mosquitoes, which could carry the parasites in their guts, and officials and researchers focused on improving mosquito control.
This included clearing swamps, setting up mosquito nets, and developing new larvicides and pesticides. These control measures would help alongside treatments like quinine, a remedy from the Cinchona tree that had been used since at least the seventeenth century and was isolated in 1820.
Hopes for a malaria vaccine, though, were faint.
Even today, parasites are seen as bad luck in vaccinology. No other vaccine has yet been developed for a parasite that causes disease in humans – although several have been made for parasitic diseases that affect livestock and pets.
This is because the complexity of parasites leads to several difficult, though not insurmountable, challenges for scientists: finding a suitable animal model, culturing the parasite in the lab, and identifying and refining the ingredients needed for a vaccine.
The first, crucial step is finding an animal model – an animal whose experience of the disease closely resembled ours. This is fundamental to learn how the pathogen infects us, causes disease, and how our immune system responds to it.
In the case of malaria, it would be an animal that could be infected by a very similar Plasmodium species (if not the exact same one), via bites by an Anopheles mosquito. Hopefully, its immune system would react to the parasite like ours does. Practically, it would be a mammal, and one that’s easy to work with in a lab. How about rodents?
But rodent models for malaria would take decades to find, and would be infected by a different Plasmodium species than we are, making comparisons less certain. The complex life cycles of parasites often make them highly adapted to each species – they respond to specific signals from the species they infect, which trigger their transformations.
These differences – between malaria in humans versus other animals – meant that even if scientists had developed a vaccine that protected animals in the lab, it wouldn’t be at all guaranteed that it would protect us too, because our infection could develop quite differently.
For example, after scientists discovered Plasmodium species that infected birds – like ducks and pheasants in the 1930s and ‘40s, they began screening many new drugs to test their potential for antimalarial effects. But several turned out to have serious side effects in humans, suggesting there were important underlying differences. Other animals like rhesus monkeys proved expensive and impractical to work with.
Faced with these challenges, many researchers turned to another option – humans.
Malaria had become a common treatment for syphilis between the 1920s and 1940s. This was because the Austrian scientist Julius Wagner-Jauregg had discovered ‘fever therapy’: that patients could be cleared of advanced syphilis if they experienced persistently high fevers, such as those caused by malaria.
Malaria fever therapy was effective because the bacteria that causes syphilis, like many others, canʼt easily survive high temperatures. So syphilis patients could be infected by malaria, and then their malaria symptoms could be treated with antimalarial drugs.
For some time, the number of syphilis patients who were treated with malaria meant research into human malaria was feasible – although not at all without risks. But by the 1940s, fever therapy was replaced by penicillin, which could now be mass manufactured and had become quickly and widely adopted.
Only in 1948 was a rodent model for malaria finally discovered.
A few years prior, a doctor named Ignace Vincke had been working on malaria control in different provinces of the then-Belgian Congo, during World War Two. Alongside his duties, he was also conducting research on local mosquitoes.
He tested the mosquitoes to see which animals they had bitten using ‘precipitin tests’ – because mosquitoes ingest antibodies from animals during their blood meal. The results were hopeful: the mosquitoes tested negative for antibodies from primates, cattle, horses and antelopes, sheep and dogs – so their target was likely another animal, potentially a rodent or insectivore.
A few years later, he returned to the region to work with Marcel Lips, an entomologist who had carried forward his research. Lips had noticed fresh parasites in the blood of local Anopheles dureni mosquitoes, but couldn’t find which animal they had fed on.
Over the next two years, they collected and tested hundreds of wild rats near the Kisanga river in the now-Democratic Republic of Congo without much luck. Part of the struggle seemed to be because of a forest fire that had deterred Anopheles mosquitoes from the area. Finally in 1948 they found one rodent – a thicket rat – that was infected by blood parasites.
After extracting the parasites from it, they managed to reproduce the malaria infection in laboratory rodents. This demonstrated that the life cycle could be completed. The newly-discovered species, now called Plasmodium berghei, was quickly shared with malaria researchers around the world, along with the Anopheles dureni mosquito.
Unfortunately it was not a perfect model, because the Anopheles dureni mosquito was fragile and difficult to breed in the lab. Other researchers abroad were unable to see it transmit the Plasmodium parasite and were limited to studying only the initial stages of the parasite – until 1964, 16 years later.
The promise of DDT
In the meantime, a major breakthrough had shaken up the field: the pesticide DDT.
In the late 1930s, Paul Müller and other scientists at the Swiss company Geigy had been synthesizing chemicals and testing whether they’d work as insecticides.
Many insecticides had been found among the thousands of organic compounds that make up coal tar. Coal tar was already known to have medical consequences – such as the cancerous effects of soot, which had been seen in chimney-sweepers.
Some constituents of coal tar (such as para-chlorinated diphenyl ether and many sulfur analogs) had become a focus for researchers in the chemical dye industry, who noticed their strong toxic effects on fabric-eating moths.
Building on this knowledge, Müller and his team took a new approach: they synthesized and screened hundreds of similar organic compounds from coal for insecticidal effects, until they finally discovered DDT in 1939.
225 parts of chlorobenzene are mixed with 147 parts of chloral or the corresponding amount of chloral hydrate and then 1000 parts of sulphuric acid monohydrate are added. Whilst stirring well the temperature rises to 60ºC and then sinks slowly down to room temperature, the mass then containing solid parts. It is poured into a great deal of water, whereupon the product separates in solid form. It is well washed and crystallized from ethyl alcohol forming fine white crystals, having a weak fruit-like odour.
– European patent GB547874A by Geigy for DDT.
Years later in the speech for his Nobel prize awarded for this discovery, Müller would describe the ideal insecticide as one that met the following criteria: It would be greatly toxic to a wide range of insects, but safe for mammals and plants; would cause no irritation and have no odor; would have a long-lasting effect; and could be manufactured at a low price.
On these qualities at least, DDT was highly effective. It worked against fleas and lice (which spread typhus), mosquitoes and flies (which spread malaria, dengue, yellow fever and dysentery), beetles, cockroaches, ants, and many more agricultural and industrial pests. DDT could also be used in a wide range of forms: powder, solutions, emulsions, and suspensions. Unlike previous insecticides, which had to be sprayed indoors on a regular weekly basis, it was stable on wall surfaces and its effects lasted for months.
Since DDT was an insect nerve poison developed during World War Two, research also went into its potential harms in humans and other animals.
As early as the 1940s, studies had found it was harmful at high doses for fish, birds, crabs, frogs, and a range of ‘beneficial insects’.
And yet, it seemed far less harmful for mammals than previous insecticides, which were based on nicotine and arsenic. Even if ingested incidentally while using it for household pest control, a large share of DDT would be stored in our fat tissues, apparently causing us little harm otherwise.
So the side effects of DDT garnered little attention at the time except among experts, though they led to usage recommendations and labellings in some regions.
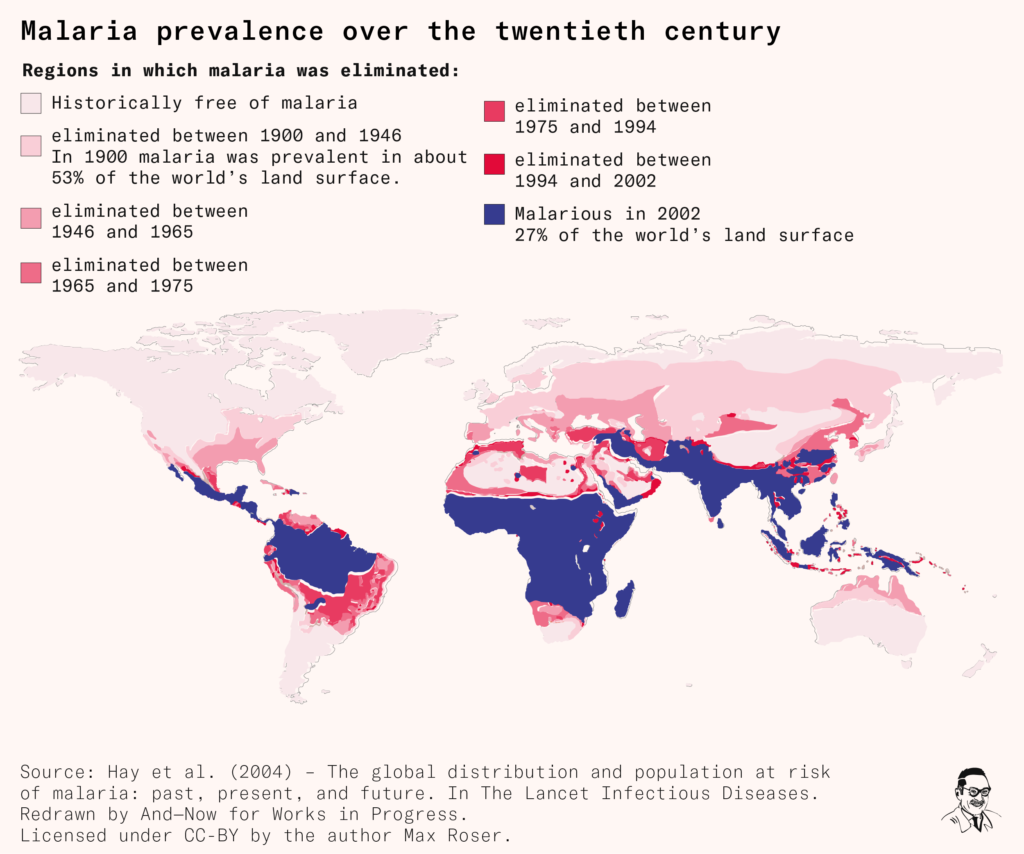
Authorities began to use it extensively, and national DDT-spraying programs in several countries such as Italy and Chile were quickly successful. Officials in Sardinia, for example, had implemented a mass spraying campaign in 1946 that eliminated malaria within four years. The implications felt historic – malaria had been endemic in Italy since at least ancient Roman times. And in 1951, the following year, malaria was also eliminated in the United States.
But a pressing problem was beginning to emerge – insects were developing resistance to DDT. This had been noticed in house flies by 1947, in the Anopheles mosquitoes that spread malaria by 1951, and across a growing number of species and regions. The biological mechanisms behind how this happened were at this point though unclear.
Worries grew that the pace of spraying wasn’t sufficiently fast or coordinated, and interest shifted towards a global strategy, which would be led by the UN and its agencies. So in 1955, the Global Malaria Eradication Program was launched by the World Health Organization.
The development of mosquito resistance to DDT in some parts of the world suggests there is no time to be lost in eradicating the parasite while it is still possible to control the vector with chlorinated hydrocarbon insecticides.
– Charles W Mayo and Frederick J Brady, the Eighth World Health Assembly, 1955
A major premise of the WHO’s program was that existing tools were sufficient, and success depended primarily on execution. So field scientists were recruited to become its operators and managers, and largely withdrew from their research. All countries were instructed to strictly follow detailed, standardized manuals of operation. Research funding by the WHO was limited; in the US, now free of malaria, research funding also contracted swiftly.
In 1956, the year after the eradication program’s launch, the United States became its biggest funder. The ‘conquest against tropical diseases’, it was argued by the US Secretary of State George Marshall and other officials, would have great economic benefits for agricultural productivity and international trade.
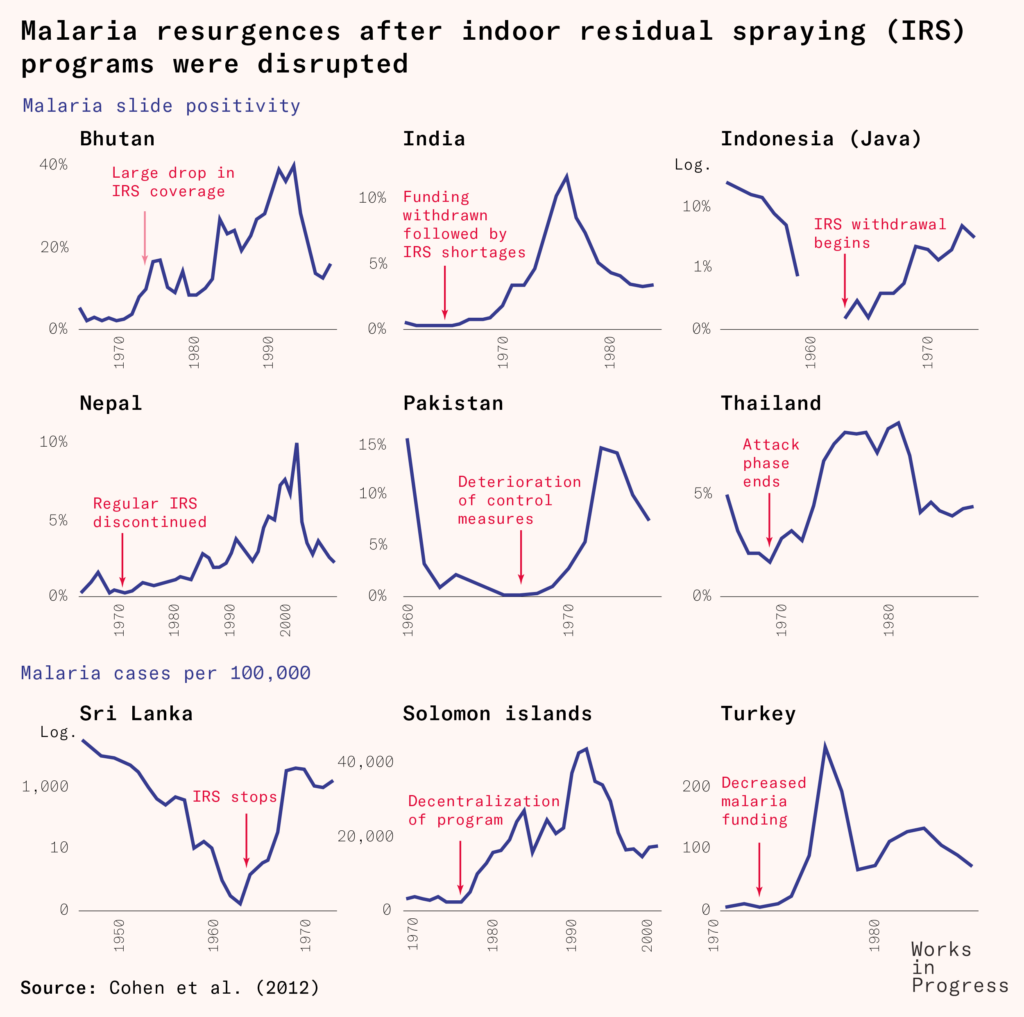
The global eradication program led to a great decline in malaria prevalence, to the point of elimination in several countries including Cyprus, Hungary, Spain, Taiwan, and Jamaica.
But the program’s greatest weapon – DDT – was gradually losing its strength, as insect resistance to it continued to evolve and spread further.
More cracks in the program began to emerge. All sub-Saharan African countries, for example, had been ‘excluded from the eradication programme, for physical, economic and developmental reasons complicated by high endemicity and prolonged transmission factors’ as the World Health Assembly stated in 1957. Previous local elimination programs hadn’t shown success, so new ones were only planned as experimental pilot programs.
Alongside these factors, there were wars, natural disasters, migrations, changes in climate and mosquito habitats, and agricultural developments that led to resurgences of the disease across Africa, South Asia and South America.
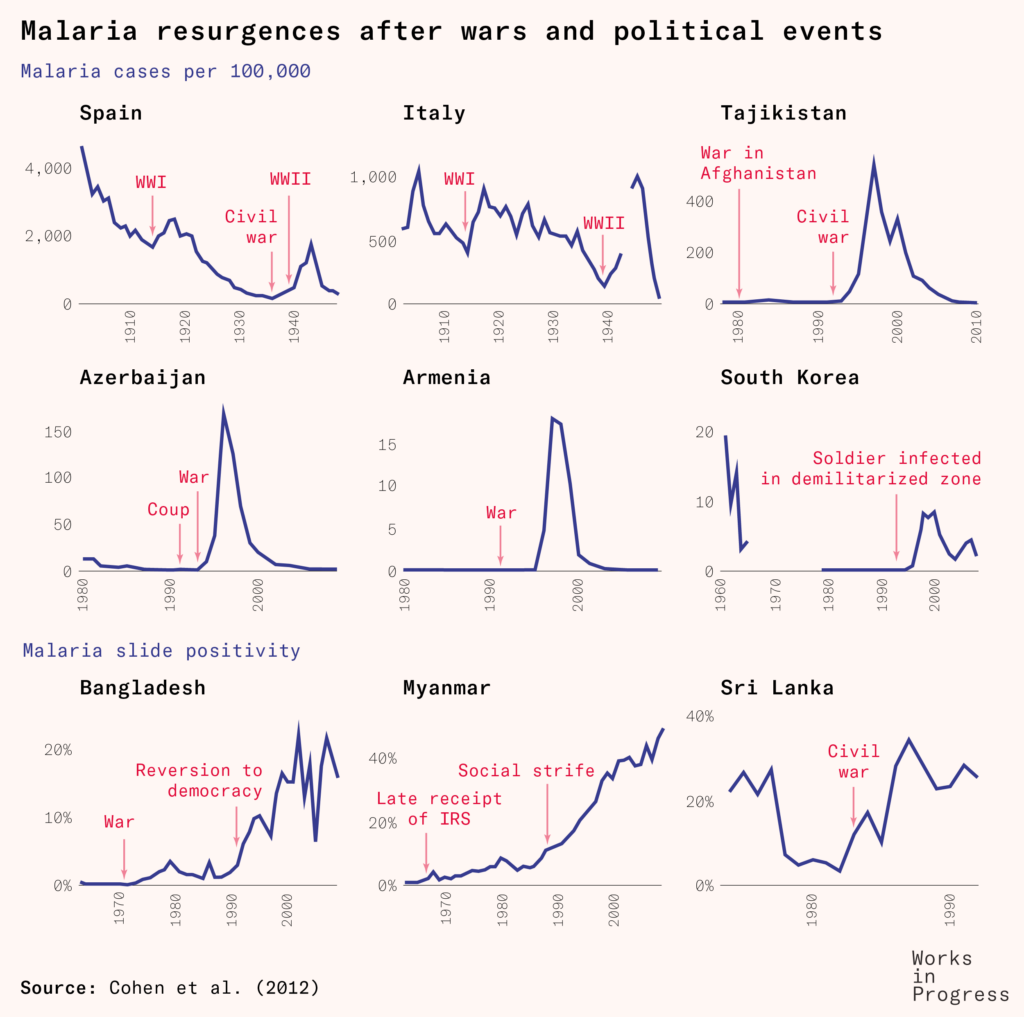
Meanwhile, recognition grew of the effects of DDT on wildlife and biodiversity: it was highly persistent in animals and the environment; it thinned birds’ eggshells and harmed fish and aquatic life.
This reached a zenith in 1962, when the marine biologist Rachel Carson’s book Silent Spring was published, massively raising awareness of the harms of mass pesticide use. Although it was highly influential, it was also seen by some experts as overdramatized. Yet additional studies substantiated several of the environmental harms it described, and these led to phase-outs and bans of the pesticide in agricultural use in many countries.
In 1963, America ended its contributions to the WHO’s Malaria Special Account, which had made up more than 85 percent of the total funding. Part of the WHO’s general budget was reallocated to bridge the gap, but the challenges of eradication continued to grow.
Local malaria control measures faced funding shortages, disruptions, withdrawals, and in several cases just complacency. The disease remained a large burden in much of the world.
Refining animal models
It was around this time, in the early 1960s, that the answer to a more practical rodent model finally emerged – which would allow researchers to carefully test new drugs and vaccines.
The malariologists Meir Yoeli and Jerome Vanderberg had immersed themselves in the original papers where Vincke and Lips had described malaria infections in thicket rats years earlier, and a detail caught their attention.
Vincke and Lips had described the typical malaria season of thicket rats (found in forests in Central Africa) as occurring in forest temperatures between 18 to 21ºC. This was much cooler than the outdoor environment at around 30ºC – which researchers abroad had been working with. So Yoeli and Vanderberg tested these cooler conditions in a lab – and they succeeded in replicating the entire life cycle of Plasmodium berghei. After the results were published in 1964, this lower temperature range became the standard used when working with the rodent model.
Vanderberg later recalled how these constraints also stemmed from the specific Plasmodium species that had been found in the rats.
If another central African parasite such as Plasmodium yoelii had been discovered first, the much more relaxed temperature requirements for infecting mosquitoes would have allowed sporogonic development to be regularly achieved in the laboratory many years sooner.
– Jerome Vanderberg, 2008
So the same step could have been sped up in multiple ways: through closer attention to the early findings; or with more resources and researchers working on the problem; or through pure chance, by stumbling upon a more practical wild parasite first.
Nevertheless, work on a vaccine could now really get going.
By this time, the US government was eager to renew malaria research, in order to protect troops fighting in the Vietnam War, who encountered drug-resistant malaria. The US army set up major research programs on new antimalarial treatments and potential vaccines.
And in 1967, the WHO called for a re-examination to the eradication strategy, switching focus from eradication to control. Across the 1970s, massive malaria epidemics affected South Asia and Turkey, and threatened resurgences elsewhere.
Altogether, organizations turned again to funding malaria control and R&D that could pay off in the long-term.
The first signs of promise
One of the key research teams included Ruth Nussenzweig, Jerome Vanderberg, and their colleagues at the US National Institutes for Health (NIH) and the Walter Reed Army Institute of Research (WRAIR).
They first tested out traditional vaccines that contained the initial ‘sporozoite’ stage of the parasite. This might not have worked: If there was even a single parasite that survived and made it to the liver, it could cause disease. So our immune system would need to react very quickly.
The team’s idea was to radiate these sporozoites with X-rays – which would weaken them until they were unable to reproduce or cause disease, but could still trigger an immune response. These weakened parasites would then be injected into mice’s veins, and then the mice would be challenged with bites from malaria-infected mosquitoes to see if they were protected.
In 1967, this culminated in the first-ever sign of promise: while 90 percent of unvaccinated mice developed malaria, only 37 percent of vaccinated mice did. They soon found the mice were protected against other Plasmodium species as well.
But translating this result into humans would be a challenge.
The sporozoite preparation came directly from the salivary glands of mosquitoes, which had been dissected, ground up, and irradiated. But how to purify this preparation remained an open question. It mattered because waste material from mosquitoes’ salivary glands could be dangerous, causing embolisms or severe reactions if injected.
So, the team decided to irradiate live, infected mosquitoes, and let them transfer the parasite to the mice naturally, through a bite.
Around the same time, they noticed something else that would prove pivotal to malaria vaccine development. Serum from some vaccinated mice formed a precipitate around the parasite, after incubation. This reaction implied that they had an immune reaction towards the parasite, and it became known as the ‘circumsporozoite precipitation’ (CSP).
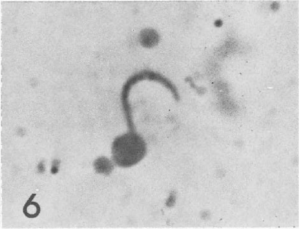
With this knowledge, the team led further successful studies in mice and monkeys, and then began to conduct ‘human challenge trials’.
Human volunteers in these trials would stay in a room as they were exposed to tens to hundreds of parasite-infected mosquitoes, which had been weakened by X-ray radiation. Then, months later, they (along with a control group who hadn’t received the earlier irradiated bites) would be ‘challenged’ several times by healthy mosquitoes carrying the parasite.
The researchers would then compare how many remained protected, and repeat the challenges over several months. Any volunteers who developed the disease would be treated with antimalarial medicine.
The first attempts to protect volunteers were unsuccessful. So the researchers increased the amount of radiation applied to the mosquitoes. In a challenge trial in 1973, they saw complete protection in one of three volunteers, across repeated challenges over seven months. His serum also showed the CSP reaction. This time, with the new dosing, the modest finding was replicated by several other research teams.
But how to scale up the concept – which had required dissecting mosquitoes – remained perplexing.
Would researchers need to breed massive numbers of mosquitoes? Then, would they need to let them feed on parasite-infected mice, radiate them, and then decapitate them, to extract the sporozoites from their salivary glands?
One company, called Sanaria, set out to work on the problem and continued for decades.
But the impracticalities led most researchers to turn to other ideas – such as vaccines against other stages of the parasite’s life cycle.
The underlying problem here, once again, was that the parasite undergoes the stages of its life cycle in different host organisms and conditions. With our current understanding and technology, this makes parasites very difficult to culture in a lab.
So why not try another strategy? The majority of vaccines used today contain only a few key antigens – rather than the entire pathogen organism – that are sufficient to stimulate the immune system.
But, since the malaria parasite was so large – with over 5,000 genes in its genome, and many proteins on its surface, which shuffle around across stages of its life cycle – the question remained: Which antigens should be included?
One possibility was worked out in the early 1980s by the same team, who continued to investigate the CSP reaction (where serum from mice had attached to the parasite). With new monoclonal antibody technology, they identified a part of this reaction: antibodies had bound to a specific protein on the parasite’s surface. It became known as the CSP protein, and they sequenced its genetic code. It appeared to be highly genetically similar across different Plasmodium species, suggesting it could also protect against a broad range of them.
Now, the concept was ready to be turned into a vaccine.
The emergence of the RTS,S vaccine
At this point, the team entered a collaboration with Smith, Kline & French (which later became GlaxoSmithKline), aiming to use their recombinant E. coli technology to produce synthetic CSP protein. They developed four candidate ‘DNA subunit vaccines’, that each contained a part of the CSP protein from the Plasmodium falciparum parasite, and first tested them in animals. Out of these, they selected one promising candidate for further testing.
In a small-scale human trial in 1986, they found that while it was safe, it did not seem very effective, with only one volunteer protected out of a group of six.
So they continued to work on further refinements. For example, it was known that ‘subunit’ vaccines, which contain only one or a few proteins of the pathogen, are often insufficient to trigger much response on their own. So the team also experimented with adding adjuvants – ingredients used to boost the immune response by making the protein more identifiable to the immune system.
Over the years, they tested more than a dozen different versions of the vaccine – with different adjuvants and formulations – in both field trials and experimental challenge trials. Most yielded disappointment.
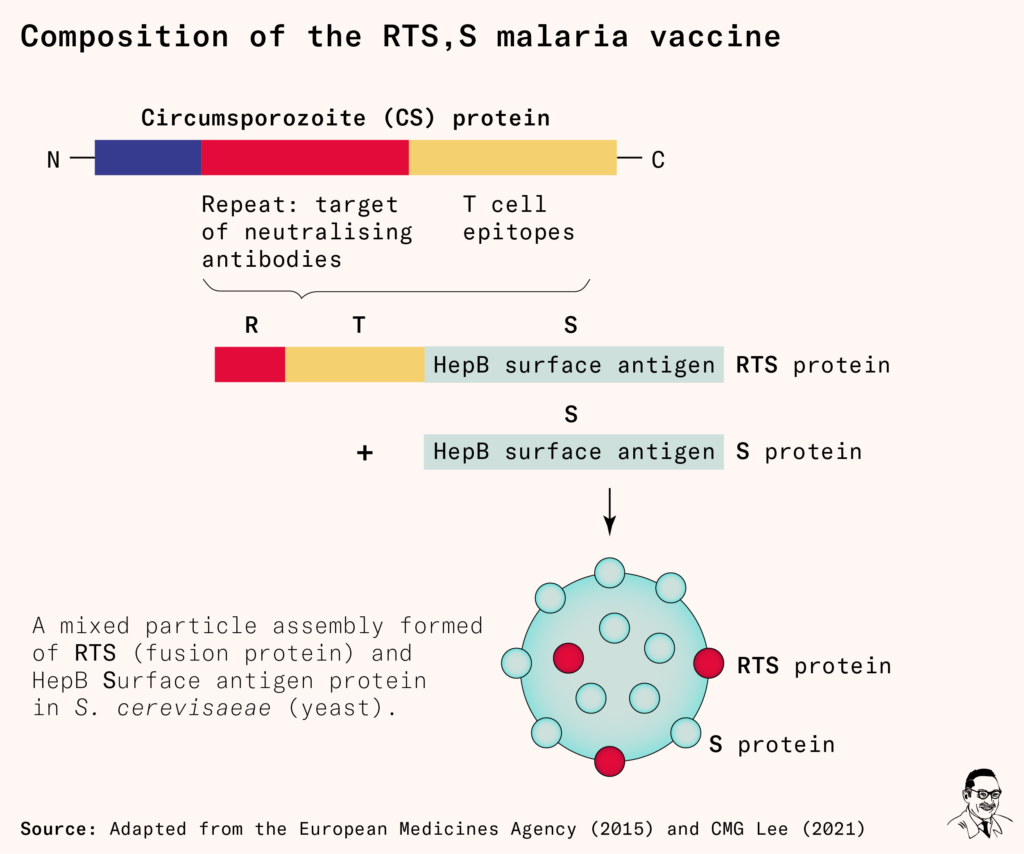
But there was one exception. It was a formulation of a new fusion protein that contained four components: the CSP protein’s repeat regions (R), T-cell epitopes (T), and an adjuvant – the hepatitis surface antigen (S). When the RTS protein and additional S protein (used as a scaffold) were produced by their yeast cell technology, the proteins would assemble into particles that resembled a virus.
The team believed this virus-like appearance would make it easier for our immune systems to recognize than the previous protein had been.
The formulation was called the RTS,S vaccine.
In a 1997 human challenge trial, this RTS,S vaccine protected six out of seven of the volunteers. Although it was only a small pilot trial, it appeared to be far more effective than any other formulation so far.
The team moved to field studies, testing the vaccine in 300 adult men in the Gambia in a phase one trial in 1998. Although the malaria vaccine was intended to be a vaccine for young children – who make up the vast majority of deaths from the disease – it’s common to perform clinical trials on healthy adults first, in case of unexpected side effects.
The results were promising. The researchers found it was broadly safe and reduced the chances of infection by 34 percent over four months.
Brian Greenwood, one of the researchers who led the trial, described the results to Undark magazine. ‘That was really the start of RTS,S’ he explained. But in his view, people’s interest in the vaccine was primarily about intellectual curiosity. ‘I don’t think there was any sort of push. It was done by people who were more academics and interested in the immunology,’ he said. ‘It wasn’t seen as a public health issue.’
But while the RTS,S vaccine showed promise, it was also far from ideal. Its efficacy began high but appeared to decline after a few months. Even after three doses, its efficacy was only 30–40 percent, averaged over the next four months.
Funding a vaccine without profit
In 1999, Ripley Ballou, a vaccine researcher on the team, met with GSK executives in Belgium to discuss the trial’s results.
We had this glimmer of hope that came from this study that says, you know, what, something is happening here, and we think we really need to take it to the next step.
– Ripley Ballou
That next step would be trials in children, who make up the majority of deaths from the disease. The situation for GSK wasn’t expected to be financially rewarding: they would eventually invest hundreds of millions of dollars in funding clinical trials for it – but even if it was approved, the market would be poor countries, and GSK committed to offer it at a not-for-profit price.
Despite the risks, GSK gave the green light to the vaccine – but only if Ballou and his colleagues could find additional funding for the project. By this time, long after the end of the Vietnam War, the US army was no longer interested in funding the vaccine – not believing it would be worthwhile or effective enough to protect military personnel.
Eventually, the team found their additional funding from the new Malaria Vaccine Initiative at PATH, which had been recently established after a grant from the Gates Foundation.
Yet, because no strong ‘correlate of protection’ had been found (that is, there was no immunological signal that predicted who had developed protection), it was difficult to demonstrate that the findings could be extrapolated from a single trial. So, to gather evidence that it was broadly effective, the researchers were to run field trials in different regions, and at a range of different dosings.
And because of worries of potential side effects, the trials ran sequentially down age groups: the vaccine was first tested in the oldest children (6–11 year olds), then in younger children (1–4 year olds), and finally in infants under one year old.
All in all, this meant the trials were much longer and more expensive than typical vaccine trials – and each part of the logistic process was hampered by funding struggles and shortfalls.
But the researchers persevered. Over seven years, between 2001 and 2008, they carried out phase one and two trials in several sites in the Gambia and Mozambique.
‘The outcome was extremely promising’, wrote Ripley Ballou in 2009, ‘not only was the vaccine extremely well tolerated and highly immunogenic – it reduced the risk of infection by 65.9 percent (95 percent CI: 42.6– 79.8 percent, P < 0.0001) during a three-month follow-up period.’
By 2009, 11 years after the first field trials in the Gambia, the vaccine was now about to begin phase three of the regulatory process, which involved more trials across several countries. These would hopefully demonstrate that the vaccine was broadly effective in different regions and demographics.
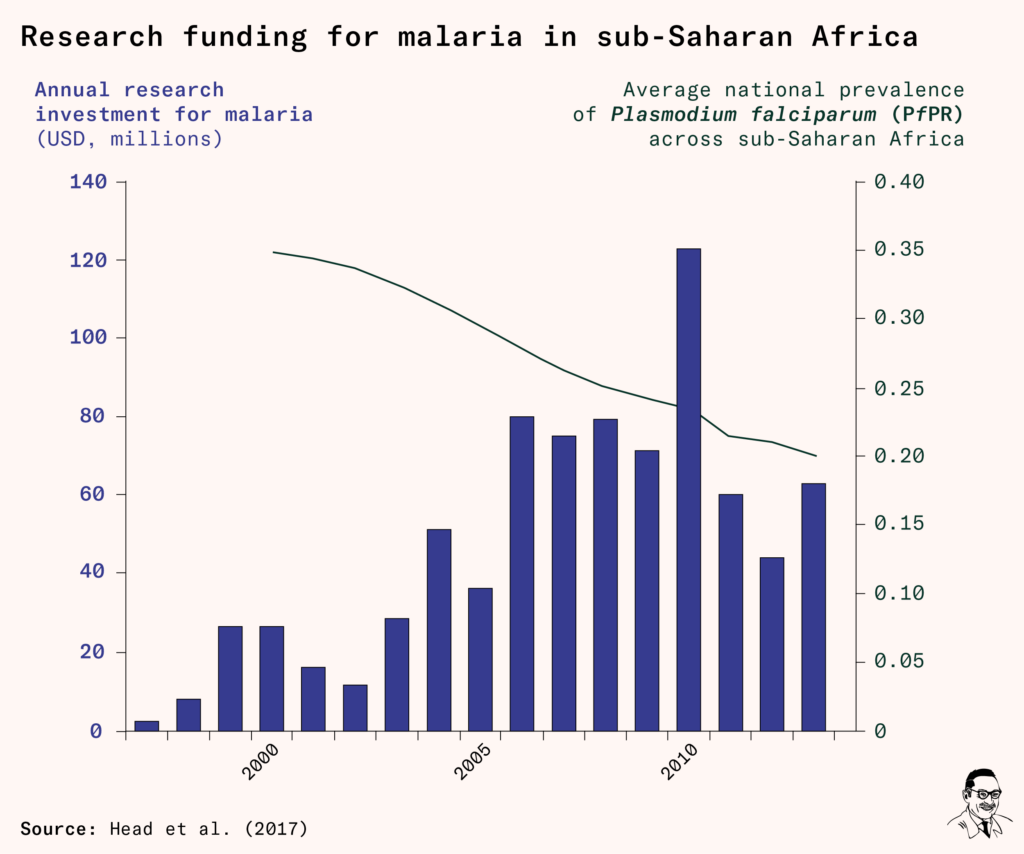
Fortunately, by this point, funding to tackle malaria had grown thanks to new initiatives like the Global Fund in 2002, the US President’s Malaria Initiative in 2005, Unitaid in 2006, and funding from the Gates Foundation. Annual spending on malaria control was around $1.78 billion, while it was $500 million for malaria research, and a smaller share, $160 million, for malaria vaccine research specifically.
Research funding temporarily rose at the end of the 2000s as funders pitched in to cover the cost of late-stage trials for the RTS,S vaccine in Africa.
The last mile
In 2015, when the phase three trials were finally complete, the results looked positive. The vaccine reduced the risk of clinical malaria by 56 percent in children aged 5 to 17 months and by 31 percent in those aged 6 to 12 weeks – although as in previous trials, this protection declined after a year.
By this time, the vaccine had already spent 17 years in clinical trials, and had cost over $550 million. For comparison, the average length of clinical trials for a vaccine is eight yearsii, and recent vaccine trials for rotavirus – which causes severe diarrheal disease in young children worldwide – have cost $100–200 million.
In July of 2015, the European Medicines Agency gave the malaria vaccine the green light, stating its safety profile was acceptable – which was a prerequisite for it to be approved by the WHO, whose decisions are used to support countries without strong regulatory bodies and recommend it for international funding.
But when the results reached the WHO, the vaccine was unexpectedly held back. The WHO recommended it only for children aged 5 to 17 months of age, and asked for further testing, pilot studies and follow-ups before it could be rolled out more widely.
‘We had to close down and put on hold the whole manufacturing side,’ said GSK’s Lode Schuerman.
Two researchers who had been involved in previous trials commented in November 2015:
As recognised by the investigators, the study includes several unproven assumptions. The most important of these is the assumption that RTS,S/AS01 will have a significant impact on mortality, a key factor for decision makers and for calculation of DALYs; this assumption is not yet supported by empirical data.
– Brian Greenwood and Ogobara K Doumbo
A second reason was that – according to speculative post-hoc analyses of the data – there appeared to be a higher rate of meningitis in older children who received the vaccine than in those who didn’t. There also seemed to be a higher rate of deaths among young girls, but not boys – even though the total number of deaths in either group was small.
The WHO explained that these cases were not actually believed to be related to the vaccine itself: they weren’t related to the timing of the doses, only occurred in patients in two of the trial sites, and included different types of meningitis. Nevertheless, they argued, the disease needed to be ruled out as a potential side effect with more pilot studies.
So now the RTS,S vaccine would undergo additional studies: in more children, in more countries. The pilot projects took four years to set up – as researchers now needed to raise additional funds and hire new staff – and finally got started in 2019.
They then faced practical challenges that were reminders of the real life impact of the vaccine. One example was the choice of locations. The project ran within specific districts of regions, leaving many parents in other districts disappointed and confused. Some traveled with their children to the eligible districts to get them a dose of the vaccine.
Another was the criteria. Some participants explained that the 15 month gap between the third and fourth doses was too long for them to remember to return to the vaccination clinic. In addition, children over six months were excluded from receiving the first dose. Health workers reported that it was difficult and painful to explain this to parents and caregivers, who were desperate to have their children protected.
In April 2021, when two years of pilot studies were finally complete, the Data Safety and Monitoring Board (DSMB) examined the results. They showed no increase in rates of meningitis, deaths, or any other signs of safety concerns.
The vaccine was finally endorsed by the WHO in October 2021 for broad use in children.
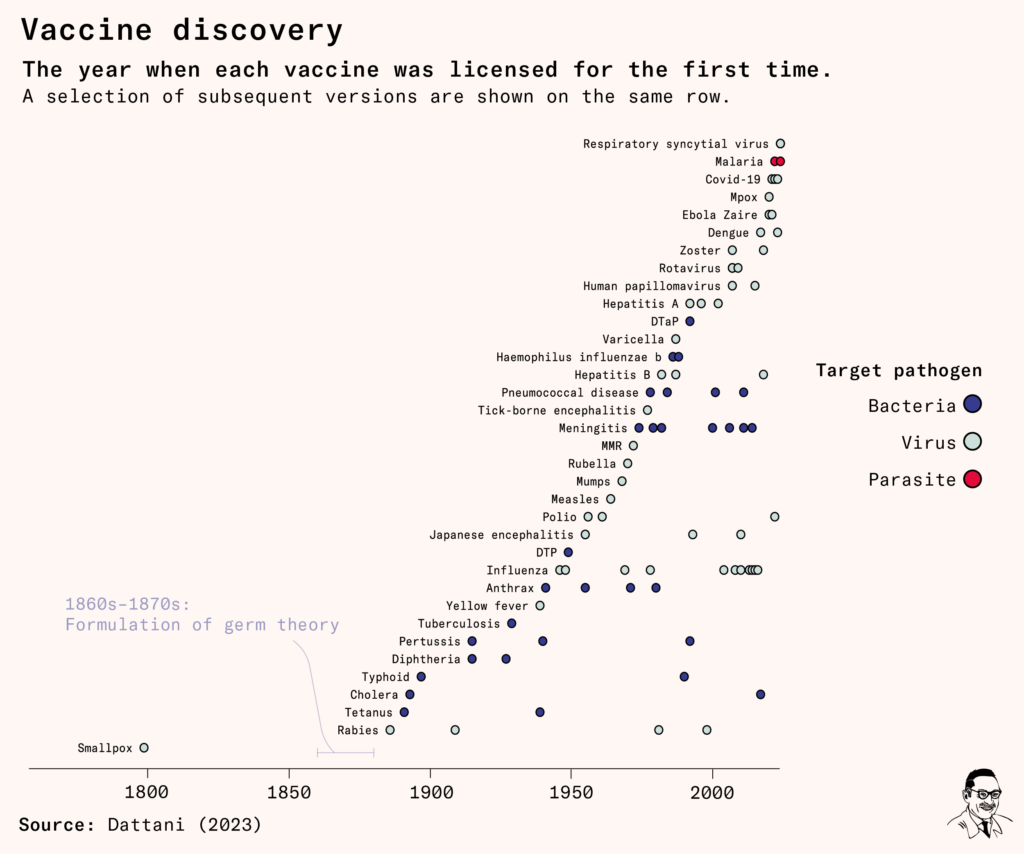
In sum, the RTS,S vaccine spent 23 years in 25 trials and pilot studies, before it was licensed.

In sum, the RTS,S vaccine spent 23 years in 25 trials and pilot studies, before it was licensed.
Since the RTS,S vaccine was approved, more than 1.2 million children in Kenya, Malawi and Ghana have received it, with support from Gavi, the vaccine alliance. 18 million more doses have been allocated over the next few years, but production is expected to ramp up slowly, because the antigen and adjuvant are in limited supply.
As of 2023, two more malaria vaccines are on track for approval. One is the R21/Matrix-M vaccine, which has already been licensed in Ghana and Nigeria after successful phase two trials, but is only likely to be approved by the WHO several years from now. It is an iteration on the concept of the RTS,S vaccine – by using the same ‘active ingredient’, the CSP protein – within a different formulation and with a different adjuvant.
The other is the PfSPZ vaccine – which came from scaling up production of the irradiated sporozoite, by breeding mosquitoes and using tiny mosquito guillotines to decapitate them and extract the parasites. It was developed by the company Sanaria and is now in late-stage clinical trials.
Why we didn’t get a malaria vaccine sooner
‘We should have had this vaccine a long time ago,’ Alassane Dicko, a professor of public health and malaria researcher in Mali who worked on some of the trials, said to Reuters.
Malaria victims are ‘not Europeans, they’re not Australians, they are poor African children,’ said Ashley Birkett, director of the malaria vaccine initiative at PATH to Undark magazine. ‘Unfortunately, I think we have to accept that that is part of the reason for the lack of urgency in the community.’
‘Primarily, this is the problem that you face when you’re trying to develop a vaccine that nobody wants to pay for,’ said Ripley Ballou.
Reports of the malaria vaccine are peppered with similar quotes from scientists and public health professionals who worked in the area.
This isnʼt the typical narrative we hear about new discoveries and technologies. We tend to think of them emerging as soon as they’re technically possible.
But vaccines are driven by people motivated to work on the problem, and need to clear significant economic and regulatory hurdles, as well as scientific ones, before they make it to the general public.
The development of malaria vaccines was stalled over and over again: by the focus on the eradication campaign and suspension of research; then by the lack of funding and urgency to address what had become a distant problem for the West; then by the complexity, length and cost of running the clinical trials; and then by the heightened and shifting regulatory requirements that added years of additional funding struggles and studies.
Perhaps the most revealing part of the story was what it didn’t cover. Why wasn’t there already enormous demand and funding to push for a vaccine against malaria – a widely-recognized disease that causes 600,000 deaths a year?
Underlying it all are challenges that are much wider than malaria.
Getting a vaccine through clinical trials is expensive for individual firms. It’s time-consuming, and typically fails.
A successful vaccine takes nine years on average to get through trials and receive approval – but 94 percent of vaccines in the pipeline have failed. And only a small number of research sites across Africa are able to perform large-scale clinical trials even today. For diseases that affect the poorest, who wouldn’t be able to afford the high prices needed to recoup the cost of developing vaccines, there is little incentive for firms to go through this costly and risky process.
Consider tuberculosis. Its vaccine, the BCG vaccine, is now almost a hundred years old. It’s been used to vaccinate children, but provides little protection for adults, who now make up the majority of deaths. Because itʼs a greater risk to people with HIV/AIDS, and those who lack healthcare, its death rate is now thirty-times higher in sub-Saharan Africa than in Europe. In total, it still kills over a million people worldwide per year.
Like malaria, developing a tuberculosis vaccine for adults has been considered challenging for decades. The Mycobacterium tuberculosis bacteria remains silent in the body for many years, which means it takes a long time to test vaccines for it in clinical trials; and we lack close animal models of the disease.
Nonetheless, after years of research, researchers have developed tuberculosis vaccines that have shown efficacy in adults in early trials – but continue to face struggles in funding. GSK, which developed the leading TB vaccine candidate, recently licensed the vaccine to the Gates Medical Research Institute to take forward trials rather than continue themselves. It’s now expected to take another four to six years to complete phase three trials, and then will need a partner for large-scale manufacturing.
The lack of strong incentives to solve these diseases makes it grueling to raise the capital to invest in vaccines, let alone with the speed and urgency that’s needed.
These problems are also why we don’t have vaccines for other tropical diseases like trachoma (the leading infectious cause of blindness)i, as well as highly fatal, sporadic diseases like Marburg (which has killed around 50 percent of known cases), Nipah (73 percent), and Crimean-Congo haemorrhagic fever (40 percent).
To get at least one vaccine successfully through phase one and two trials – for each of 11 priority infectious diseases – researchers at the Coalition for Epidemic Preparedness Innovations (CEPI) have estimated it would cost between $1.2 to 8.7 billion. Compared to the burden of disease, and their risks of spreading further, the returns to society for making such investments would be huge.
How do we do it?
Cultivating new vaccines
Typically, innovation is incentivized through patents. This is when innovators are given a temporary monopoly: an exclusive right to sell products using their innovation. Policymakers donʼt have to choose which ideas to support before the research happens – instead, companies that make successful discoveries and find a market for their innovations are rewarded.
The temporary monopoly means that firms can command a higher price than they would in a market where anyone could produce the drug after it had been discovered. Typically, firms recoup the costs of R&D by selling at a high price in rich countries. Then, once the product is off-patent or new competitors emerge, prices come down and the drug or vaccine gets used more widely.
For diseases that mainly affect the poor, however, firms can’t recoup their costs in rich country markets, and they might not be able to charge high enough prices to generate large enough returns to attract enough commercial investment.
In sub-Saharan Africa, the region most affected by malaria, only around USD $73 is spent on healthcare per person per year – which is 80-times smaller than what’s spent in wealthy countries. But even within poor countries, it’s the even-poorer rural population, who are less able to spend on healthcare, that suffer the most from disease.
The presence of aid agencies and international institutions doesn’t necessarily solve the problem either. As large buyers, these institutions have a lot of bargaining power – Gavi, for example, sources 90 percent of vaccines in Africa. Such institutions might try to bargain prices down after firms have already invested in R&D and manufacturing capacity.
Anticipating this, firms are more reluctant to invest in the first place. In economics, this is known as the ‘hold-up problem’. Similar problems often arise between utility companies and power generators in markets with a single utility company – for example, when power generation firms are reluctant to invest in expensive power plants, knowing the electricity utility company can hold the price it offers down once the investment is made.
In sum, the market rewards for developing vaccines for diseases such as malaria are often too weak to attract enough commercial investment, especially given the risk of failure.
And even if firms could charge high prices for vaccines for diseases like malaria, it would not be economically efficient to do so. With high prices, lots of individuals and countries wouldn’t be able to afford to pay for the high price, and millions wouldn’t benefit from the new vaccine.
Both the economically-efficient and the just solution is to make sure that firms get rewarded for investing in vaccine development – but not through higher prices.
This is crucial because the benefits of tackling malaria by getting a vaccine out to all those at risk are immense.
Malaria’s burden is enormous – it is the fifth-most common cause of death across Africa, and the Global Burden of Disease study estimates that per year, it costs around 46.4 million disability-adjusted life years (DALYs, a standardized unit to calculate years lost or lived with disability from different diseases). Even if people were willing to pay only $100 to avoid one DALY, an effective vaccine would be worth billions of dollars per year. For context, the UK’s healthcare system spends about £30,000 per DALY saved, and US payers tend to be willing to pay about $50,000-60,000 per DALY saved.
How do we align the commercial incentives for vaccine innovation with their social benefits? How do we make it profitable to do the right thing – without limiting access by charging high prices?
If we could crack this problem, it would go a long way to overcome challenges across the whole process.
Push funding or prizes?
The usual approach to addressing this market failure is for donors to provide funding upfront, through grants. This is called ‘push funding’ because it involves paying for inputs.
Push funding is often useful when you know who is best placed to develop the innovation, or want to fund open-ended basic research. But it comes with risks – you have to pay even if researchers fail to deliver. And, importantly, it doesn’t on its own help ensure that the final product reaches all the people who need it.
Push funding is how malaria vaccines have been funded so far – with around $160 million per year from philanthropic donors and industry since 2007 – but this is far short of the billions of dollars per year an effective vaccine could be worth.
Push funding also comes with pitfalls. It means bureaucrats need to choose which ideas to bet on. But bureaucrats may not be good at predicting which ideas will be successful, and researchers and firms may have an incentive to exaggerate their prospects. Companies working on fixed contracts also lack ‘skin in the game’, and receive funding whether they succeed or fail. Even academic researchers have reasons to stray from the task, devoting effort to preparing their next grant application or incentives to work on unrelated projects that will advance their careers.
Another idea is to use prizes instead. These could provide a reward for making a solution available for a specific problem – such as developing a malaria vaccine that meets a minimum level of efficacy and high standard of safety.
The best use of a prize is when people are rewarded for developing an idea that anyone can copy. But copying vaccines is hard. Even after a vaccine was developed, it still wouldn’t be easy for another company to reproduce it, even if the hurdle of intellectual property rights were waived. This is because there are details that are difficult to capture in documentation, called ‘tacit knowledge’ – like specifics about the biological processes and very new technologies used in manufacturing. In other words, we don’t just want a vaccine to be invented – we also want to have it scaled up.
Advance Market Commitments
Enter Advance Market Commitments. A standard Advance Market Commitment (AMC) is a promise to subsidize the future purchase of a new vaccine in large quantities – if it’s invented – in return for the firm charging customers close to marginal cost (that is, with only a small mark-up).
Letʼs break it down. The subsidy incentivizes research by compensating innovators for their fixed cost investments in R&D and manufacturing capacity. The commitments to buy a certain quantity at a certain price ensure the vaccine is affordable and widely available. The subsidy is conditional on a co-payment (this is the part that is close to marginal cost) from governments in low and middle income countries – without it, the developer receives nothing. This incentivizes firms to develop vaccines countries will actually use, not just those that meet technical specifications.
So while patents trade-off innovation incentives with affordable access, AMCs help us achieve both. And the price strategy means that AMCs encourage deployment at scale in a way that most prizes do not.
AMCs are a kind of inversion to typical ‘push funding’ – they instead ‘pull’ innovation towards a goal by paying for outputs and outcomes. They don’t require funders to choose which research efforts to back in advance – they can just commit to rewarding the innovations that succeed. And they’ve been successful at doing so in the past.
One of the earliest proposals for AMCs was for malaria vaccines, made in the 2004 book, Strong Medicine by one of us – Rachel Glennerster – and Michael Kremer. If it successfully fostered an effective vaccine, it was estimated that a USD $3.2 billion AMC, which committed to pay $13–$15 per person for the first 200 million people immunized, would have saved disability-adjusted life years for less than $15 each – an extraordinarily cheap intervention.
At the time, malaria vaccines were considered innovations that were far out of reach. In hindsight, we can see that they weren’t as infeasible as people feared.
This was a missed opportunity, because AMCs are suited to targets that may seem out of reach, but might not actually be – if the target is not met, the funder does not have to pay, so there is little downside to offering one for an ambitious target.
The reason we missed this opportunity may be because policymakers and pharmaceutical companies have a bias against ambitious targets, in favour of ‘quick wins’. Companies encourage funders to pay for near-term targets which they are more confident they can meet.
An AMC is intended to cover a company’s fixed R&D and capacity costs. Funders will err on the side of being generous to incentivize the firm’s participation (given the large social benefits) – they may defer in their price and subsidy setting to companies working on the problem who are better informed but also have an incentive to exaggerate costs. This is less true for far-off targets where both sides are relatively in the dark about the costs of developing the innovation.
These factors may be why there was more support for an AMC for pneumococcal disease than malaria. A new pneumococcal vaccine for strains in poor countries was an important, but near term target. Pneumococcal disease killed around 800,000 young children each year. Vaccines already existed, but didn’t yet protect against the bacterial strains that were found in poor countries. New vaccines to address them were in late-stage clinical trials in the 2000s, but lacked a big enough financial market to make it to market, or at least to do so quickly.
So in 2009, several countries – Italy, the United Kingdom, Canada, the Russian Federation and Norway – and the Gates Foundation launched an USD $1.5 billion AMC for new pneumococcal vaccines. An economics expert group specified the details – the subsidy, the co-pay price, and the supply commitment to incentivize firms to produce at scale.
One by one, three pneumococcal vaccines were approved and met the criteria: one by GlaxoSmithKline (in 2010), another by Pfizer (in 2010) and a third by Serum Institute of India (in 2020).
Supplies of the pneumococcal vaccine rose rapidly, as did its uptake: in countries eligible for Gavi support, vaccination rates converged to global levels five years faster than it did for the rotavirus vaccine – which was also funded by Gavi, but without an AMC. By 2021, the three pneumococcal conjugate vaccines were estimated to have saved 700,000 lives.
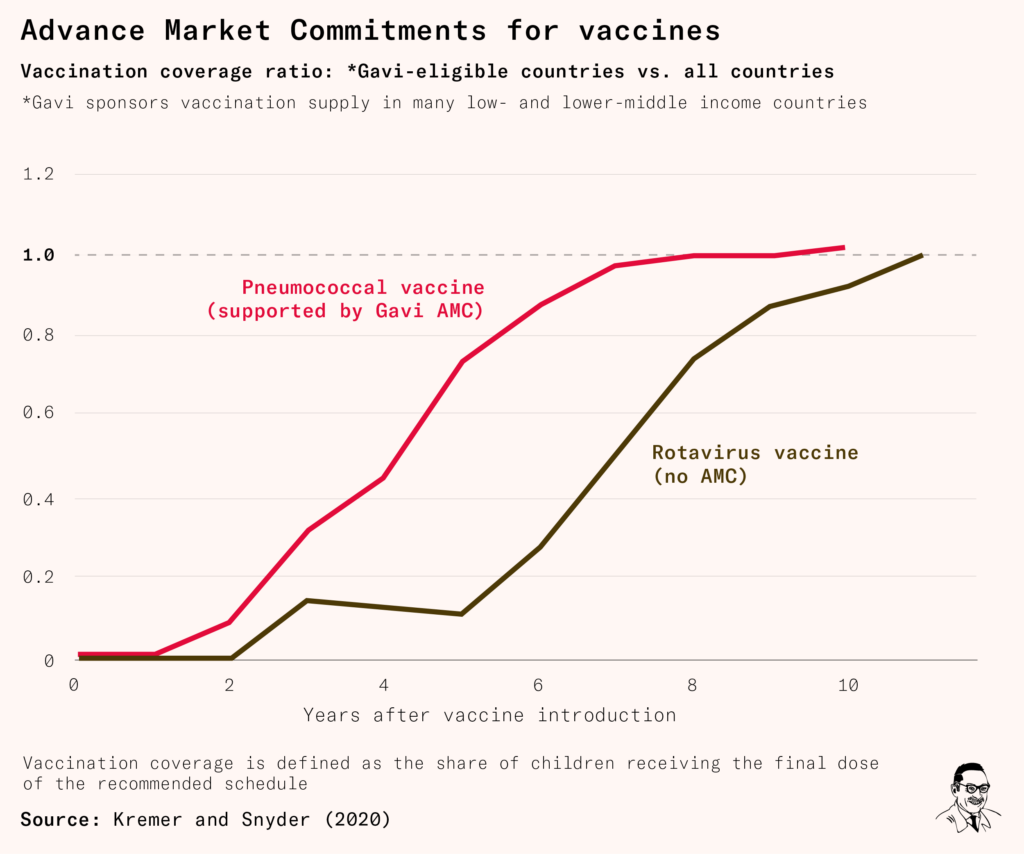
It’s likely that AMCs would have sped up the development of a malaria vaccine, but they can still help now – to develop vaccines for tuberculosis, trachoma, Zika, Nipah and many other diseases.
The arrival of a second malaria vaccine illustrates some of the tradeoffs involved in incentivizing innovation. Once a vaccine is developed, it is easier to develop alternatives. For one thing, there is now proof it is possible, making it a much less risky undertaking. If a ‘copycat’ vaccine which free rides on the R&D investment done by others manages to grab most of the vaccine market and most of the AMC subsidy, then this undermines the incentive for the first firm to do the hard work of inventing the first vaccine.
One answer is only to allow the first firm to market with a vaccine to get any of the AMC subsidy. But this causes its own problems: if two firms are independently investing in a new vaccine and one gets to market faster by one day, they should not capture all the AMC as this would generate too much uncertainty for those investing in vaccines. For the patients’ sake we also want to buy the best vaccine on the market, not the one that came first.
This trade-off is also relevant to policy conversations about incentivizing the new generation of coronavirus vaccines. One answer is to reward the best vaccine that meets the criteria within a certain time window, say six months. The window should be long enough to ensure firms who are doing genuinely separate research are rewarded and have the incentive to take a bit longer to get a better product, but it should also be short enough to prevent me-too or copy cat vaccines capturing the gains.
Advance Market Commitments are one approach to overcome the barriers, but there are also other parts of the process that can be improved: the organization and infrastructure of clinical trials, basic research into parasites and their immunology, adjuvants and vaccine platforms, and closer models for a range of diseases.
The future
The world now has a malaria vaccine. Although it’s not perfect – and malaria control efforts with bednets and treatments will continue to be vital – itʼs an undeniable breakthrough. But it’s one that took far too long. Each part of the journey to develop, refine, test and approve vaccines was delayed by funding struggles, while more than half a million children were dying from the disease each year.
Not every pathogen we’ll face will have such a complex life cycle or lack close animal models. But the overarching problem – the lack of funding to develop vaccines for the global poor – will remain. Millions of lives can be saved by solving it.
With Advance Market Commitments, we can draw out effective vaccines and scale them up rapidly. For what amounts to small sums to rich countries, we can stimulate research that will have enormous benefits to health and wellbeing around the world, and de-risk and hasten the process to find and reward the most promising ideas.
From a scientific perspective, it’s never been easier to develop new vaccines. We can now see pathogens on a nanoscopic level, test and sequence their genomes rapidly and cheaply, boost our immune response with adjuvants we hadn’t discovered before, and deliver vaccines with safer technology. The question isn’t necessarily whether we can make breakthroughs, but when and what’s needed to get there.
It may have taken us 141 years to get a malaria vaccine, but it doesn’t have to take anywhere near as long for the next one. We can start making up for lost time.